



Mobile Detection developement (Tallinn University of Technology)
The main research in the last six months (from October 2014 to April 2015) was focused on the following:
- The optimization (amount of dervatizating agent, derivatizating temperature and heating time) of derivatization procedure of sulfur mustard hydrolysis and oxidation products (thiodiglycol (TDG), TDG sulphoxide (TDGO) and TDG sulphone (TDGOO))
- The optimization of the separation conditions (buffer pH and concentration, capillary temperature and applied voltage).
- Validation of capillary electrophoretic separation method with direct UV detection (linearity, precision, LOD and LOQ).
- Preliminary studies of sediments (spiking of sediments with TDG, TDGO and TDGOO and evaluation of adsorption to the sediment; separation of pore water from sediment)
1) Optimization of derivation procedure
To obtain reproducible results from the sample derivatization procedure [1], a careful optimization of several reaction parameters, such as the amount of dervatizing agent, the heating time and heating temperature was performed. The effects of the amount of the derivatizating reactant was added, were examined. Each time sample was injected into an electrophoretic system, the average peak area (n =3) was measured and the condition that gave maximum response (peak area) was selected. Based on the stoichiometry of the derivatizing reaction, the required minimum amount of reactant was calculated (~40 µL per 2.5mg of TDG). Then 2-, 3- and 4-fold volume excess of reactant was applied for the derivatization of each analyte. Based on the average peak area measurements, the maximum response was achieved applying the 2-fold volume excess of phthalic mixture. The appearance of the phthalic acid peak on an electropherogram (unreacted phthalic anhydride) acted as an indicator of a sufficient excess of derivatizing reactant. For future experiments, 100 µL of reactant per each 2.5 mg of analyte was selected to avoid the lack of derivatizing reactant in real samples.
To find the optimal derivatization temperature and heating time, a set of additional experiments was carried out, varying the temperature in a range of 25-85◦C and the heating time in a range of 0-25 minutes. Keeping the temperature constant (85◦C, in accordance with the literature source [1]), the maximum response was achieved within 5 minutes (Fig.1A). The effect of the heating temperature was evaluated, keeping the reaction time (20 min) constant. The best response was obtained at 45◦C and further temperature increases did not affect the result (Fig.1B). Finally, the derivatization conditions were as follows: the amount of reactant – 100 µL of reactant per each analyte, derivatization temperature 45◦C and derivatization time 20 minutes.
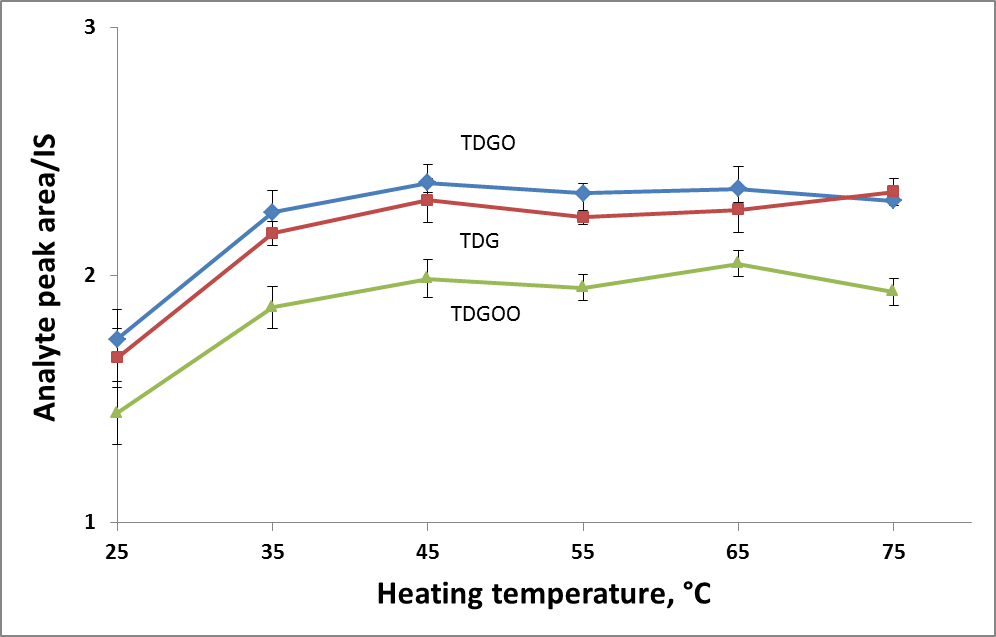
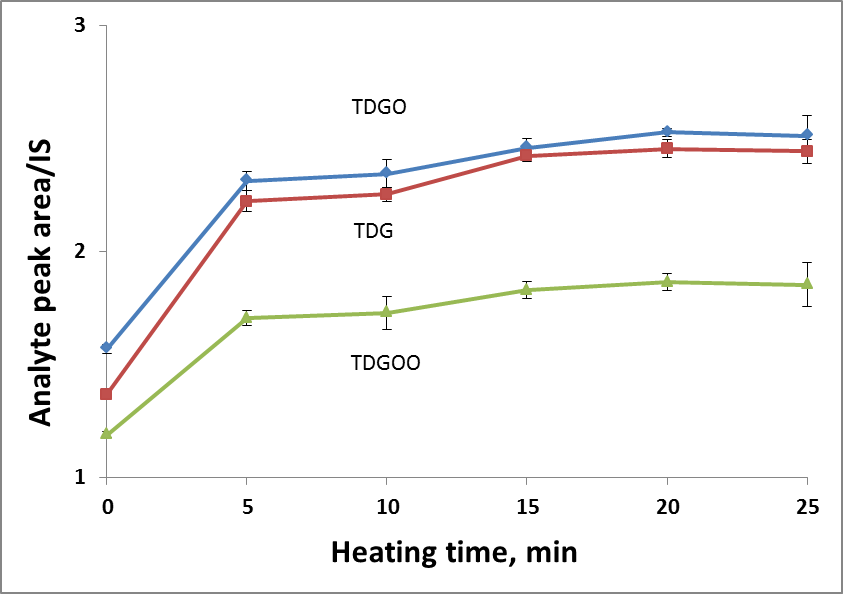
Figure 1. Effect of heating temperature (A) and time (B) on derivatization efficiency
2) Choice of background electrolyte
Underivized TDG and its oxidation products are neutral at pH below 9 and, thus, can be analysed using the MEKC separation technique. Applying 50 mM borate buffer with 100 mM SDS and direct UV detection at 200 nm, TDG and TDGO could be separated within 7 min. The LOD for TDG and TDGO was 0.1 mM. The high values of LOD are logically justified due to the absence of strong UV adsorbing sites. In this work, an improved method for the analysis of target sulphur compounds involved derivatization with phthalic anhydride incorporating chromophore sites into the analyte structure and, at the same time, affecting the pKa value of the formed derivatives and giving a charge to the molecules at pH below 9.
The optimization of the separation conditions was carried out by the investigation of the effect of buffer pH, concentration, capillary temperature and applied voltage on separation efficiency.
It is well known that buffer pH plays the key role in optimization of a separation process affecting the EOF velocity and degree of analyte ionization. So, in the present work a 30 mM borate buffer with a pH range of 7.5 – 10.0 was investigated to evaluate the impact on separation results. At buffer pH values below 8.0, the derivatized analytes were unresolved and the peak shapes were asymmetric. An increase in the buffer pH value to 8.5 led to improvement in analysis time and all analytes were baseline separated. Further pH change to 9.5 resulted in an increase in the electrophoretic mobility of the analytes. Baseline separation was still achieved and the analysis time was the shortest. Unfortunately, starting at pH 9.0, the sulphone peak area was prone to rapid decrease and at pH 10 almost disappeared, which indicated a strong addiction of thiodiglycol sulphon stability from the pH value of the separation media.
The influence of borate buffer concentration on separation was studied in the range of 20-50 mM at pH 8.5. It was expected that by increasing the buffer concentration the migration time of each derivate would also increase (almost double). Nevertheless, the best separation efficiency was achieved at 30 mM and further concentration increase led to peak broadening without a remarkable change in peak separation.
Additionally, the effect of the applied voltage over the range 15-25 kV and capillary temperature (15-30 C) was also investigated in terms of separation efficiency and migration times. Voltage values above 20 kV resulted in faster migration times of analytes, but the separation efficiency was not sufficient. All derivatives were baseline resolved at 15 kV, but the analysis time was extended by several minutes. The increase in capillary temperature also noticeably improved the migration time of the derivatives. Thus, the temperature increase from 15 to 30oC decreased the analysis time by a quarter, keeping the separation efficiency at a reasonable level.
Finally, the optimized separation conditions for the separation of three derivatives were as follows: borate buffer concentration 30 mM, buffer pH 8.5, applied voltage 15 kV and capillary temperature 25 °C. The representative electropherogram is shown in Fig. 2.
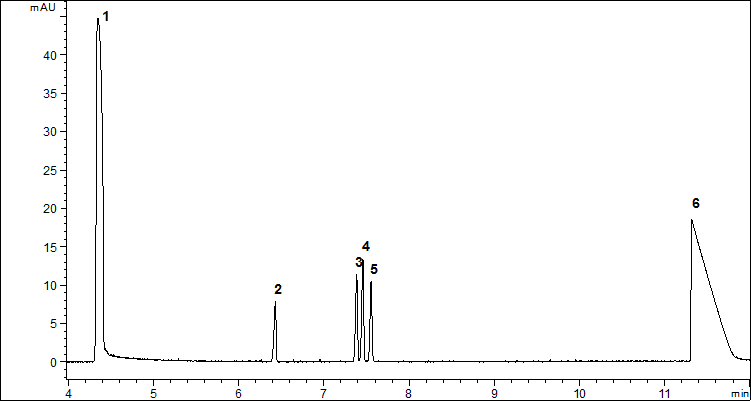
Figure 2. Representative electropherogram obtained after derivatization of 3.05 µg/mL of TDG (4), 3.45 µg/mL of TDGO (3) and 3.85 µg/mL of TDGOO (5) under optimized separation conditions: borate buffer concentration 30mM, buffer pH 8.5, applied voltage 15 kV and capillary temperature 25°C. Additional peaks: EOF (1), IS (2) and phthalic acid (6)
3) CE method validation
The precision of the developed method was investigated. All precision tests were based on optimized background electrolytes and a standard mixture of derivatives. The tests were performed for the run-to-run and day-to-day variations of the migration times and peak areas. Run-to-run precision resulted in relative standard deviation (RSD) values of 0.6% (n=6) and 2.4-3.1% (n=6) for the migration time and peak area, respectively. Additionally, day-to-day results showed RSD values of 1.2% (n=6) for the migration times and 3.6% (n=6) for the peak areas. There were no systematic changes in peak shape during the precision tests.
The linearity was evaluated in the range of 0.10 – 2.44 µg/mL for TDG, 0.14-2.76 µg/mL for TDGO and 0.15-3.08 µg/mL for TDGOO. Calibration curves were constructed using 5 concentration levels and were based on the ratio of the corresponding derivative to IS peak area versus concentration. The linearity range, regression equations and regression coefficients are shown in Table 1.
Table 1. Regression data for the calibration curves
|
Derivative |
Linearity range µg/mL |
Regression equation |
Regression coefficient, R2 |
Limit of detection, ng/mL |
Limit of quantification, ng/mL |
|
TDG |
0.10-2.44 |
y=0.1517x + 0.0017 |
0.9996 |
98 |
305 |
|
TDGO |
0.14-2.76 |
y=0.1359x – 0.0161 |
0.9997 |
138 |
415 |
|
TDGOO |
0.15-3.08 |
y=0.1233x – 0.0304 |
0.9994 |
154 |
463 |
The limit of detection (LOD) and quantification (LOQ) were obtained experimentally by measuring the signal-to-noise ratio (S/N). The lowest LOD and LOQ were obtained for TDG and calculated as 98 ng/mL (S/N=3) and 305 ng/mL (S/N= 10), respectively.
4) Preliminary studies of sediments
The sediment samples were collected from the bottom of the Baltic Sea at the port of Virtsu (Läänemaa, Estonia; Shceme 1) by the Marine System Institute at TUT. The collected sediment samples were divided into aliquots and stored in a freezer.

We assume that these samples do not contain the degradation products of sulphur mustard.
The sediment samples were spiked with TDG, TDGO and TDGOO water solutions (final concentration 100, 200 and 400 µM of each). Then the samples were homogenized and shaken for 4 h, after that the samples were placed into refrigerator for 20 h and 7 days.
At the beginning of sample preparation, the sample was taken out from the refrigerator, and left to stand at the room temperature for 1 h. Afterwards the sample was centrifuged for 30 min at 6000 rpm, excess pore water was collected and the remaining sediment was weighed. Then the acetonitrile was added to the sediment sample and the sample was shaken for 20 min at room temperature. Next the sample was centrifuged for 30 min at 6000 rpm and acetonitrile was collected.
The collected pore water was concentrated by using powdered carbon aerogel to analyse the content of TDG, TDGO and TDGOO [2]. Acetonitrile was used for the elution of compounds of interest from carbon aerogel. Both acetonitrile fractions (from pore water and sediment) were evaporated to dryness and allowed to the derivatization and analysis procedure as described above. Results for TDG and TDGO (total incubation time 24 h) are represented in Table 2.
Table 2. Contents of TDG and TDGO in spiked sediment.
| TDG initial, µM | TDG find in pore water, µM | TDG find in sediment, µM |
|
0 |
0 |
0 |
|
100 |
10 |
10 |
|
200 |
23 |
45 |
|
400 |
81 |
138 |
| TDGO initial, µM | TDGO find in pore water, µM | TDGO find in sediment, µM |
|
0 |
0 |
0 |
|
100 |
10 |
18 |
|
200 |
76 |
30 |
|
400 |
129 |
75 |
It can be seen from the Figure 3 that the adsorption of TDG and TDGO to the sediment is rather similar.
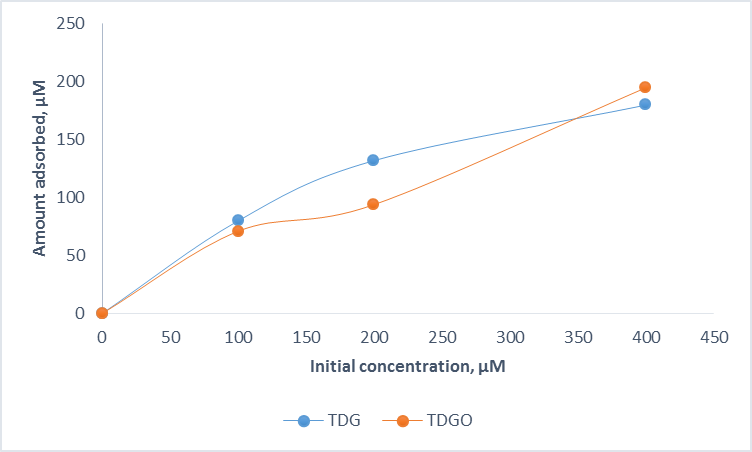
Figure 3. Adsorption data in sediment.
Based on the results of preliminary tests it can be concluded that after the incubation in the refrigerator for 7 days the TDG is completely lost, the concentrations of TDGO and TDGOO are decreased 8 and 3 times, respectively.
The investigations will be continued.
- Vanhoenacker, G., De Keukeleire, D., Sandra, P. J. Sep. Sci. 2001, 24, 651-657.
- P. Jõul, H. Lees, M. Vaher, E.-G. Kobrin, M. Kaljurand, M. Kuhtinskaja. Electrophoresis, 2015, DOI: 10.1002/elps.201500038.